Risque athérogène ou infarctus
Introduction
L'excès de cholestérol dans l'alimentation peut induire une pathologie appelée athérosclérose.
La physiopathologie de cette maladie est la suivante: le cholestérol en excédent se dépose dans la paroi artérielle. Le diamètre de l'artère diminue. Il y a aussi une calcification qui réduit l'élasticité des artères et enfin le collagène sous endothélial est mis à nu ce qui provoque des thromboses. Ce phénomène est particulièrement dangereux si l'artère irrigue un organe vital (cSur, cerveau).
Le flux du cholestérol dans l'organisme est assuré par des lipoprotéines qui transportent le cholestérol de l'intestin vers le foie ou les artères ou bien des artères vers le foie.
Pour évaluer le risque athérogène (risque de thrombose), on dose les différentes lipoprotéines et on compare leurs quantités. Si les lipoprotéines qui transportent le cholestérol vers les artères sont nombreuses, le risque est élevé. Ce risque est atténué par la présence de lipoprotéines qui assurent le retour du cholestérol vers le foie.
Lipoprotéines
a) Structure
Les acides gras et les lipides jouent un rôle fondamental dans le fonctionnement des êtres vivants : ils sont les composants principaux des membranes cellulaires, jouent un rôle dans la transmission nerveuse et certains lipides sont utilisés comme précurseurs d'hormones.
Une grande partie de ces lipides est apportée par l'alimentation. Ils sont distribués par voie sanguine et lymphatique à tous les organes du corps humain.
|
Un lipide est un
ester (combinaison d'un acide gras et
d'un alcool). Il possède donc dans sa formule chimique
de longues chaînes carbonées peu ou pas polarisées.
Cette structure explique l'effet hydrophobe des lipides
: ceux-ci sont insolubles dans l'eau . Or, le sang est
le tissu qui contient la plus grande proportion d'eau
de tout le corps humain. Comment un milieu aqueux pourrait-il transporter des molécules hydrophobes (insolubles dans l'eau ) ? |
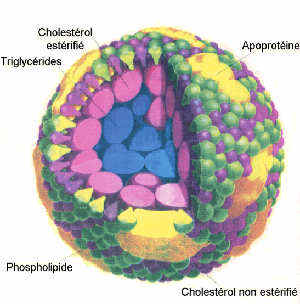
La solution réside dans la présence dans le sang de complexes macromoléculaires appelés lipoprotéines. Ces lipoprotéines sont constituées d'une association lipide-protéine. Ces protéines sont constituées d'acides aminés (AA) dont le résidu peut être très hydrophile. Dans le cas de certains AA le résidu peut même être ionisé (aspartate, asparagine, glutamate, glutamine, lysine, arginine). Ces AA présentent une grande affinité pour l'eau qui est une molécule polaire.Les lipides, en association avec ces protéines (appelées apolipoprotéines ou plus simplement, apoprotéines) peuvent ainsi être aisément transportées dans le sang.
La disposition spatiale des divers composants des
lipoprotéines est déterminée par la polarité de ces
composants : plus une molécule est polaire, plus elle sera
située à la périphérie de la lipoprotéine c'est à dire en
contact avec l'eau. Par contre, les molécules hydrophobes se
retrouvent au centre.
Les lipides des lipoprotéines:
Dans une lipoprotéine, on trouve:
- des phospholipides
|
Ce sont des esters d'acide phosphorique et de diglycérides. Ils se situent à la périphérie de la lipoprotéine. Leur partie hydrophile est tournée vers l'extérieur. Les phospholipides sont d'importants éléments structuraux de la membrane cellulaire. Ils n'interviennent pas directement dans les manifestations cliniques des dyslipoprotéinémies. |
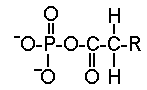
|
- des triglycérides
|
Formule générale : Leur caractère franchement apolaire les relègue au centre des lipoprotéines. Les triglycérides semblent n'être pas directement un facteur de risque cardio-vasculaire. Toutefois, une hypertriglycéridémie pourrait constituer un fait aggravant d'autres facteurs de risque reconnus (alcool , tabac, diabète...). |
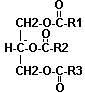
|
- Le cholestérol non estérifié (CL)
A cause de sa fonction alcool faiblement hydrophile, le cholestérol libre se situe à la surface de la lipoprotéine. Dans le règne animal, le cholestérol est le stérol le plus abondant et aussi le plus important au plan métabolique en tant que précurseur des hormones stéroïdes (progestérone, aldostérone, cortisol et cortisone, testostérone, oestradiol et oestrone). L'hypercholestérolémie est la cause de la genèse de l'athérome artériel.
- Le cholestérol estérifié (CE)
La fonction OH est liée à un acide gras supprimant ainsi tout caractère hydrophile. Le CE se situe donc au centre de la lipoprotéine.
-
Acides gras non estérifiés
- Les apoprotéines :
Comme nous l'avons vu,ces apoprotéines ont un rôle structural important. Elles se situent à la surface des lipoprotéines grâce à leurs AA hydrophiles. La partie hydrophobe de ces protéines est tournée vers l'intérieur.
Ces apoprotéines ont une deuxième fonction : certaines jouent le rôle de liguant, d'autres d'activateurs ou d'inhibiteurs enzymatiques (Voirsection c, le métabolisme des lipoprotéines).
|
CARACTéRISTIQUES PHYSICO-CHIMIQUES ET FONCTIONS DES APOLIPOPROTéINES MAJEURES |
||||||||
| Apoprotéines | Acides aminés | Poids moléculaire | Lieu de synthèse | VLDL | LDL | HDL |
concentration plasmatique (g/l) |
Fonction |
| apo A1 | 245 | 23 300 | foie, intestin | 4% | 67% | 1,00 à 1,20 | activateur LCAT. Efflux de cholestérol | |
| apo AII | 77x2 | 17 000 | foie, intestin | 22% | 0,4 | structure des HDL | ||
| apo AIV | ? | 45 000 | intestin | 0,15 | efflux de cholestérol | |||
| apo B100 | ? | 540 000 | foie | 35% | 90% | 0,70 à 1,00 | sécrétion des VLDL. ligand du récepteur LDL | |
| apo B48 | ? | 275 000 | intestin | 0,03 à 0,05 | sécrétion des chylomicrons | |||
| apo C1 | 57 | 7 000 | foie | 0,04 à 0,06 | activateur LCAT in vitro | |||
| apo C2 | 78 | 8 900 | foie | 40% | 5-9% | 0,03 à 0,05 | activateur LPL, VLDL, HDL | |
| apo C3 | 79 | 8 700 | foie | 0,12 à 0,14 | inhibiteur LPL | |||
| apo D | 169 | 33 000 | gonades, rein , foie, placenta, intestin | 0,06 à 0,07 | métabolisme des esters du cholestérol | |||
| apo E | 299 | 38 000 | foie, intestin, surrénales, macrophages | 13% | 0,03 à 0,05 | ligand du 'récepteur LDL' et du récepteur des chylomicrons résiduels | ||
b) Classification :
Il existe plusieurs sortes de lipoprotéines. On peut classer celles-ci selon :
- leur composition chimique (quantité et nature des lipides et des protéines).
- leur réactivité chimique vis à vis des polyanions et des lectines (Réaction de précipitation).
- leur réactivité immunologique vis à vis d'immuns sérums spécifiques (sérums anti-apoprotéines)
- leur origine ou devenir métabolique
- leur pouvoir pathogène (pathogénie des lipidoses)
- leurs propriétés physiques (dimension, mobilité élèctrophorétique , densité )
A l'heure actuelle, le classement est basé sur la densité des lipoprotéines par ultracentrifugation de flottation.
On distingue les lipoprotéines suivantes :
| Lipoprotéines | Densité (g/ml) |
| chylomicrons | <0> |
| VLDL | 0,94 à 1,006 |
| LDL1 (IDL) | 1,006 à 1,019 |
| LDL2 | 1,019 à 1,063 |
| HDL1 | <1> |
| HDL2 | 1,063 à 1,125 |
| HDL3 | 1,125 à 1,21 |
| VHDL | 1,21 à 1,25 |
| Lp(a) | 1,055 à 1,085 |
Plus la fraction protéique est importante, plus la
densité sera élevée.
Toutefois, les lipoprotéines de haute densité (HDL), par
exemple, n'ont pas la même composition lipidique que les
lipoprotéines de basse densité. Cependant, ce n'est pas la
densité des lipoprotéines qui nous indique le rôle de
chacune. En effet, les lipoprotéines séparées en fonction de
leur densité sont hétérogènes et leurs métabolismes très
différents au sein d'une même catégorie.
Nous avons vu que les apoprotéines jouent un rôle important
dans le métabolisme de ces lipoprotéines.
Il serait donc plus judicieux pour évaluer le risque
athérogène de baser la classification sur la teneur en
apoprotéines. Toutefois, il y a quand même une
certaine corrélation entre la fonction et la densité des
lipoprotéines.
c)Métabolisme
Le cholestérol a deux origines chez l'homme : une origine
exogène (alimentation) et une origine endogène
(biosynthèse).
- Cholestérol endogène :
La biosynthèse du cholestérol est, chez les vertébrés, microsomale. Elle est régulée par un mécanisme de rétroinhibition par un métabolite du cholestérol (le 25 hydroxycholestérol) au niveau de la Bêta-hydroxy-bêta-méthyl-glutaryl-stérol coenzyme A réductase. Cette rétroinhibition n'est jamais totale pour permettre la synthèse de polyisoprénoïdes importants pour d'autres métabolismes comme les dolichols. En effet, un précurseur du cholestérol peut également être métabolisé en dolichol.
Le foie est l'un des principaux sites de
synthèse mais toutes les cellules du corps
humain peuvent synthétiser le cholestérol. Cependant, dans
les conditions physiologiques, l'apport exogène du
cholestérol hépatique aux divers tissus est suffisant pour
que la synthèse endogène dans ces tissus soit inhibée.
- Cholestérol exogène :
Au cours d'un repas, des matières grasses riches en triglycérides sont totalement ou partiellement hydrolysées sous l'action de la lipase pancréatique en acides gras, glycérol, mono et diglycérides. Le glycérol et les acides gras libres à courte chaîne (jusqu'à 10 atomes de carbone) vont aller vers le foie par la voie sanguine (en particulier par la veine porte). Tous les autres acides gras à longue chaîne ainsi que les mono et diglycérides servent à la reconstitution de triglycérides au niveau de la muqueuse intestinale.
Le cholestérol alimentaire subit une première transformation dans les cellules de l'intestin, les entérocytes, où il est estérifié par l'acyl-coenzyme A transférase (ACAT). Dans les entérocytes, le cholestérol estérifié est associé à des triglycérides, à des phospholipides et à diverses apoprotéines dont l'APO B48 pour former de grosses lipoprotéines désignées sous le nom de chylomicrons. L'APO B48 est appelée ainsi car sa masse moléculaire est 48% celle de l ' APO B100.
Les APO C sont cédées par les HDL aux chylomicrons. Les APO C existent sous trois formes: Cl, C2, C3. Ces APO C ont peu de rôle structural et leur activité est surtout métabolique :
- La forme C1 active la lecithine-cholestérol acyltransferase.
- La forme C2 est activatrice de la lipoprotéine lipase.
- La forme C3 inhibe cette même lipoprotéine lipase.
Ce sont principalement des APO C2 que les chylomicrons acquièrent .
L'APO E est un ligand pour le récepteur hépatique à APO E.
Les chylomicrons gagnent rapidement la circulation lymphatique puis la circulation générale par l'intermédiaire du canal thoracique. Là, ils subissent l'action de la lipoprotéine lipase (LPL) attachée à l'endothélium vasculaire des muscles et du tissu adipeux. Cette action est rendue possible, rappelons-le, par l'APO C3. L'action de la LPL confère aux chylomicrons une demie-vie très brève (30 minutes) ce qui fait qu'on ne les trouve dans le sang qu'après un repas. Cette enzyme hydrolyse les triglycérides des chylomicrons en mono et diglycérides ainsi qu'en acides gras qui sont utilisés pour la production immédiate d'énergie ou mis en réserve.
Parallèlement, sous l'action d'une protéine de transfert spécifique (CETP),le cholestérol estérifié est transporté vers les particules résiduelles formées lors du métabolisme des chylomicrons, appelés chylomicrons remnants. Il s'agit là d'un échange puisque les triglycérides qui restent dans les chylomicrons sont, en même temps, transférés aux lipoprotéines de haute densité.
Il y a également un échange d'apoprotéines : nous avons vu que les HDL cédaient des APO C et E aux chylomicrons. Ces chylomicrons libèrent des phospholipides et des APO A qui sont récupérés par les HDL.
Les APO E des chylomicrons se lient ensuite aux récepteurs APO E du foie et le cholestérol est récupéré par les hépatocytes. C'est la fin des chylomicrons.
Hors des périodes de digestion, les lipoprotéines synthétisées par le foie contenant l'APO B100 transportent à nouveau le cholestérol dans les tissus. L'APO B100 est un ligand des récepteurs à LDL ou encore appelés récepteurs B,E ou bien récepteurs de Goldstein et Brown.
La majorité de ces lipoprotéines sont de grosses particules de très faible densité (VLDL) contenant en plus des APO B100 et des APO C et E. Ces VLDL, riches en triglycérides, subissent l'action dans les tissus périphériques de la LPL et de la CETP. Leur densité augmente. Elles deviennent des lipoprotéines de densité intermédiaire (IDL) contenant APO B et E. Puis, sous l'action d'une triglycéride lipase hépatique, des LDL ne contenant plus que de l'APO B. Cependant, une partie de ces IDL se lie au foie par des récepteurs BE et libèrent leur cholestérol dans les hépatocytes.
Mécanisme de dépôt du cholestérol
a) La paroi vasculaire
La structure de la paroi artérielle de la couche la plus interne à la couche la plus externe comprend :
- l'intima, formée de cellules endothéliales
- la média, constituée de cellules musculaires lisses et de tissu conjonctif intercellulaire comprenant quatre types de macromolécules: l'élastine, le collagène, les protéoglycanes et des glycoprotéines.
- l'adventice, faite de tissu conjonctif et de tissu adipeux.
b) Pourquoi une lipoprotéine est-elle athérogène ?
Au cours de la vie, un certain nombre de mécanismes vont créer des brèches dans la paroi artérielle, en particulier dans les zones de bifurcation :
- l'hypertension artérielle qui, outre son action mécanique sur la paroi, modifie le flux intracellulaire des lipoprotéines.
- les substances vasomotrices (angiotensine, catécholamines) qui arrivent à mettre à nu le collagène sous endothélial.
- les substances hypoxiantes, telles que la nicotine, qui entraînent une souffrance cellulaire conduisant à des écartements des jonctions intercellulaires les lipoprotéines qui, en cas d'élévation importante, sont capables à elles seules de provoquer une lésion.
Ces brèches vont permettre le passage dans la paroi artérielle de lipoprotéines de petite taille: HDL (6 à 14 nm de diamètre) et LDL (15 à 25 nm). Par contre, les VLDL (30 à 70 nm) et les chylomicrons (100 à 1000 nm) ne peuvent s'infiltrer.
Si la concentration plasmatique en LDL est trop élevée, il se produit une accumulation importante de lipoprotéines dans la paroi artérielle. Cette élévation de concentration peut être due à un défaut de catabolisme des LDL. Ce défaut pourrait âtre secondaire à une anomalie de l'APO B d'origine génétique ou à une diminution du nombre de récepteurs hépatiques du fait d'un apport trop important de cholestérol et de lipides saturés dans l'alimentation.
Au contact des différentes cellules présentes au
niveau de ces parois, les LDL sont alors oxydées,
reconnues par des récepteurs spécifiques situés sur les
macrophages et internalisées dans les cellules qui vont se
charger en cholestérol et progressivement se transformer en
cellules
spumeuses .
c) Oxydation des LDL
Cette oxydation est soit enzymatique soit radicalaire. Elle ne concerne que les acides gras insaturés.
Oxydation radicalaire :
Un radical libre est une espèce neutre ou chargée qui possède
un électron célibataire.
Sur le plan énergétique, les radicaux libres ont
tendance à acquérir une certaine stabilité en retrouvant un
nombre pair d'électrons. Ceci peut être réalisé de deux
manières :
- soit par le gain d'un électron comme, par exemple, pour le radical hydroxyle qui est donc un oxydant
- soit par la perte d'un électron comme pour le radical dioxyde de carbone qui est donc un réducteur.
Ces radicaux libres sont les produits réactions physiologiques. Mais ces espèces instables sont un danger pour l'intégrité des cellules, d'où l'existence d'un système de défense pour les neutraliser au fur et à mesure de leur formation.
Au cours du vieillissement, ce système de protection devient moins efficace : il y a un excès de radicaux libres dans les cellules et en particulier dans les cellules de la paroi artérielle .
Les radicaux libres vont agir avec les acides gras insaturés
des LDL et de la membrane plasmique selon
le processus de péroxydation.
Cette péroxydation aboutit d'une
part à l'établissement de liaisons parasites
protéines-lipides, d'autre part à
des désordres membranaires entraînant des
modifications de perméabilité et de fluidité de ces
dernières.
La liaison lipide-protéine influe sur la conformation et la
fonction des protéines qui n'accomplissent plus leur
fonction. La lipidopéroxydation des acides gras aboutit à la
formation de nombreuses substances: cétones, alcools, esters,
alcanes, et en particulier des aldéhydes comme le
malondialdéhyde (MDA). Ce MDA (que son origine soit
membranaire ou lipoprotéinique) serait impliqué dans la
malonylation des groupements aminés (lysine) des APO B100 des
LDL. L'APO B100 transformée n'est plus reconnue par le
récepteur membranaire spécifique de cette protéine, mais par
un autre récepteur situé sur les membranes des cellules
spumeuses ou scavenger qui sont surtout des macrophages. La
lipoprotéine est internalisée dans ces cellules
spumeuses.
d) Oxydation enzymatique :
Les lipides des LDL peuvent aussi être oxydés par une
lipo-oxygénase : la cholestérol-oxydase est une
flavoprotéine transformant le cholestérol (5-cholestène 3
bêta ol) en cholesténone (4-cholestène 3 one) par une
oxydation de la fonction alcool du carbone 3 suivie d'une
isomérisation de la double liaison (5-6) vers la position
(4-5). L'action de la cholestérol-oxydase sur le cholestérol
génère à la fois la cholesténone et le péroxyde d'hydrogène.
Le cholestérol libre des LDL, contrairement au cholestérol
libre des membranes peut âtre facilement oxydé par la
cholestérol-oxydase .
Les LDL traitées par la cholestérol-oxydase se lient de
manière beaucoup plus importante aux macrophages que les LDL
natives (fixation augmentée de 80%) et le taux de cholestérol
intracellulaire est augmenté de 51% par rapport au taux
observé avec les LDL natives.
Après internalisation des LDL oxydées, la cholesténone intracellulaire exerce une action inhibitrice sur la synthèse du cholestérol, induisant par un phénomène de rétrocontrôle une augmentation de la production de récepteurs membranaires aux LDL oxydées et donc une augmentation d'incorporation de ces lipoprotéines oxydées. A un stade où l'oxydation des lipoprotéines est encore modérée, les lipoprotéines induisent la synthèse, dans les cellules endothéliales, de molécules mobilisatrices (facteurs chémotactiques) qui attirent les monocytes circulants. Cas derniers adhèrent alors aux cellules endothéliales, puis passent entre deux cellules endothéliales pour atteindre le sous endothélium où ils se différencient en macrophages.
L'augmentation du nombre et du volume de ces cellules spumeuses réduit le diamètre des artères : La plaque d'athérome est l'endroit où se dépose le cholestérol. A ce niveau , l'artère voit son diamètre se réduire et le tissu fibreux est envahi par le calcaire. Il en résulte un épaississement et un durcissement de la paroi artérielle qui rend la circulation plus difficile et augmente le travail du cSur.
Nous avons également vu que les membranes des cellules épithéliales étaient lésées par la lipidopéroxydation. Il s'ensuit une lyse cellulaire qui met à nu le sous endothélium. Il va y avoir formation d'un thrombus selon le processus suivant : La formation de ce thrombus est favorisée par le ralentissent de la circulation dû à la diminution du diamètre des artères. En effet, il y a une accumulation de cellules sanguines et en particulier de plaquettes au niveau de la plaque d'athérome.
La Lp(a) est une lipoprotéine particulière. Selon le critère taille et composition lipidique elle ressemble aux LDL. Elle s'en distingue cependant par une composition en sucres qui lui est tout à fait particulière, à savoir un enrichissement très important en hexoses et en acide sialique. Elle renferme de l'APO B100 ainsi qu'une protéine qui lui est spécifique, l'APO (a). Cette APO (a) est reliée à l'APO B100 par l'intermédiaire d'un pont disulfure.
La Lp(a) présente une grande analogie avec le plasminogène : Le plasminogène est formé par une succession de 5 kringles appelés K1, K2, K3, K4 et K5. Un kringle est une structure en boucle. Cette structure est définie par la présence de 3 ponts disulfures intramoléculaires permettant le maintient de cette structure en cercles. Le site protéasique en position C terminale permet le passage du plasminogène à la plasmine, par l'intermédiaire de l'activateur tissulaire au plasminogène .
Pour l'APO (a), la structure est formée d'un enchaînement de 37 copies du kringle 4. Les kringles 1, 2 et 3 sont absentes. Une copie du kringle K5 est terminée par un site protéasique différent de celui du plasminogène avec présence d'une sérine à la place d'une arginine la rendant résistante vis à vis de l'activateur tissulaire au plasminogène.
La liaison disulfure entre l'APO (a) et l'APO B100 se fait par l'intermédiaire de deux cystéines au 36eme Kringle.
Le nombre de copies du K4 est variable (de 13 à 37) d'où
l'existence d'isoformes de masse moléculaire très différentes
(entre 300 000 et 800 000 daltons). On distingue 6 isoformes
:
F (faster, plus mobile à l'élèctrophorèse que l'APO B100), B
et S1, S2, S3 et S4 (S comme slower). Ces isoformes sont sous
la dépendance d'allèles qui contrôlent la nature et la
quantité de l'expression.
Rôle de la Lp(a)
Beaucoup de preuves épidémiologiques montrent que la Lp(a) est une lipoprotéine pro-athérogène et cela pour 4 raisons:
- la Lp(a) a une affinité très faible pour le récepteur BE, récepteur d'épuration des lipoprotéines. Cette faible affinité s'explique par le fait que l'APO(a), par sa liaison avec l'APO B100 diminue son affinité pour les récepteurs BE .
- La Lp(a) a par contre une affinité très grande pour les gangliosides et les protéoglycanes de la paroi artérielle. Ceci est du à l'analogie de structure de l'APO(a) avec le plasminogène. De plus, cette analogie va conduire à une compétition avec le plasminogène qui aura pour effet de baisser lefficacité de la fibrinolyse.
- La Lp(a) joue un rôle négligeable dans le transport inverse du cholestérol total .
- La Lp(a) peut se retrouver dans la paroi après oxydation et captation par les macrophages.
e) Le transport inverse du cholestérol
Les HDL sont un mélange de deux types de particules
lipoprotéiques : celles qui contiennent les APO AI et
AII (Lp AI:AII) synthétisées par le foie, et celles qui
contiennent l'APO AI mais pas l'APO AII (Lp AI) et formées
dans l ' intestin et le foie. La Lp AI possède une enzyme, la
lécithine cholestérol acyl transférase (LCAT), activée par
lAPO A1 et lAPO AIV.
La liaison de cette Lp AI aux récepteurs des HDL déclenche
l'émission d'une série de signaux cellulaires (avec
activation d'une protéine kinase C). Ces signaux provoquent
la migration du cholestérol intracellulaire vers la membrane,
permettant ainsi son exocytose. La prise en charge du
cholestérol membranaire se fait par la LP AI et la Lp AII et
est indépendante de la liaison du cholestérol à un récepteur
cellulaire. Le fait que la Lp Al:AII ne puisse pas activer le
récepteur HDL est dû aux APO AII qui inhibent l'action du
récepteur en se fixant dessus.
Les Lp AI et Lp AI:AII transportent ensuite le cholestérol vers le foie, seul organe où il pourra être catabolisé et éliminé par la bile dans les intestins.
Evaluation du risque athérogène
Que doser pour évaluer le risque athérogène?Plusieurs paramètres ont été dosés sur deux populations: une population saine et une population considérée comme "coronarienne".
Pour chaque test biologique sont comparés moyenne et écart-type relatifs aux deux populations. L' importance du chevauchement de ces deux distributions, exprimé en pourcentage, permet d'évaluer lintérêt de chaque test: Plus le chevauchement est important, moins le paramètre biologique est significatif. Inversement, un faible chevauchement conduit à la notion de paramètre hautement significatif ou discriminant.
On remarque que le caractère le plus discriminant est le dosage de l'APO B. Toutefois, l'aspect du sérum, le dosage du cholestérol total et des triglycérides restent des tests de base pour l'exploration lipidique et gardent tout leur intérêt.
Par contre, l'évaluation du risque athérogène nécessite d 'autres tests :
- Les uns quantifient directement le risque en s'intéressant au cholestérol HDL ou mieux encore , aux phospholipides HDL.
- Les autres apprécient directement le risque athérogène grâce au dosage du cholestérol LDL et de l'APO B.
Par sa fonction,l'APO B est un marqueur des lipoprotéines concernées par le risque athérogène et les APO A sont des marqueurs des lipoprotéines antiathérogènes. Il est donc intéressant de faire un rapport B/A pour évaluer le risque athérogène.
Toutefois, il faut faire attention à toute augmentation de l'APO B qui n'est pas obligatoirement une augmentation des LDL mais une augmentation des VLDL: les hyperLDLémies et les hyperVLDLémies ne se traitent pas de la même manière. Les triglycérides ne participent pas directement à l'athérogénèse mais une forte triglycéridémie rend le sérum opalescent et plus visqueux, ce qui ralentit le flux sanguin. Le thrombus peut donc se former plus facilement.
Nous avons déjà vu que seule une concentration élevée en
cholestérol LDL corrélée à une alimentation riche en
triglycérides suffisait à déclencher le processus de
l'athérogénèse. En effet, le foie n'est plus en mesure
d'éliminer l'excès de cholestérol. Celui-ci se dépose donc
dans la paroi artérielle. Mais il existe également des
hyperlipémies athérogènes d ' origine génétique.
a) Type IIa ou hypercholestérolémie
familiale
Physiopathologie :
On considère que l'hypercholestérolémie familiale résulte
d'une déficience de récepteurs cellulaires LDL se
matérialisant soit par l'absence de ces récepteurs
(récepteurs "négatifs" chez certains homozygotes), soit par
la non-reconnaissance de ces récepteurs (récepteurs «
déficients").
Cette anomalie conduit à l'accumulation de LDL circulantes
riches en cholestérol et à leur épuration par un mécanisme
anormal responsable du dépôt lipidique au niveau de la cornée
et de la paroi artérielle.
De plus, l'absence de récepteur LDL supprime l'inhibition de
l'HMG CoA réductase conduisant à un excès de synthèse
périphérique du cholestérol non estérifié qui renforce le
processus pathologique.
b) hyperlipémies de type IIb
La physiopathologie de cette maladie est mal connue.
c) Hyperlipémie de type III
Il y a une accumulation d'IDL résultant d'un blocage enzymatique intravasculaire empêchant le passage des VLDL aux LDL. Le diagnostic de certitude repose sur le rapport cholestérol VLDL/triglycérides. Si ce rapport est inférieur à 0,30 et s'il y a en APO EIII, on peut diagnostiquer une hyperlipémie de type III.
Par David Puech
Technicien de laboratoire
| Source : |

|

|
#COVID-19 : le point de situation épidémiologique sur le coronavirus SARS-CoV-2
Descripteur MESH : Lipoprotéines , Risque , Cholestérol , Hypercholestérolémie , Infarctus , Métabolisme