
Maladie d’Alzheimer : les biomarqueurs redessinent le diagnostic sans effacer la clinique
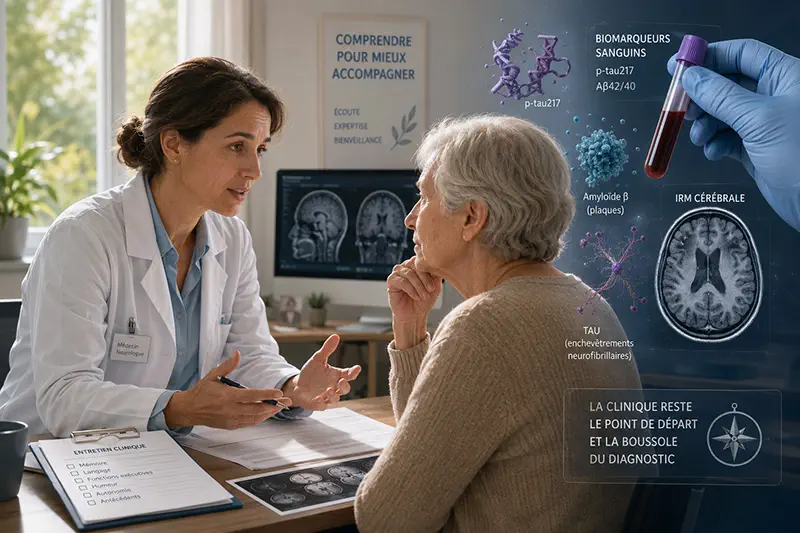
Longtemps définie par les troubles de la mémoire, le déclin cognitif et la perte progressive d’autonomie, la maladie d’Alzheimer est désormais aussi caractérisée par les mécanismes biologiques qui la précèdent et l’accompagnent. L’imagerie cérébrale, l’analyse du liquide cérébrospinal et les biomarqueurs sanguins, notamment la p-tau217, rapprochent le diagnostic d’une médecine plus individualisée.
Cette évolution ouvre la voie à une détection plus précise et à des traitements ciblant les dépôts amyloïdes. Elle soulève cependant des interrogations sur la fiabilité des tests, leur accessibilité, l’annonce diagnostique, le risque d’effets indésirables et la pertinence clinique des bénéfices observés.
À retenir — lecture rapide
- La maladie d’Alzheimer représente 60 à 70 % des démences, sans se confondre avec leur ensemble.
- Les critères de 2024 distinguent la maladie biologique de son expression cognitive et fonctionnelle.
- Les biomarqueurs sanguins peuvent orienter le diagnostic, mais ne remplacent pas l’évaluation clinique.
- Le lécanémab et le donanémab ralentissent modestement le déclin chez des patients très sélectionnés.
- En France, la HAS a refusé l’accès précoce puis le remboursement de ces deux traitements.
Une maladie fréquente aux contours épidémiologiques encore incertains
En 2021, 57 millions de personnes vivaient avec une démence dans le monde et près de 10 millions de nouveaux cas survenaient chaque année, selon l’Organisation mondiale de la santé (OMS). La maladie d’Alzheimer en constitue la cause la plus fréquente et contribuerait à 60 à 70 % des cas.[1]
Cette proportion ne doit toutefois pas être assimilée à l’ensemble des démences. Les atteintes vasculaires, les maladies à corps de Lewy, les dégénérescences frontotemporales et les formes mixtes occupent également une place significative, particulièrement aux âges avancés.
En France, l’Institut national de la santé et de la recherche médicale (INSERM) estime que 1 à 1,2 million de personnes vivent avec la maladie d’Alzheimer et que 225 000 nouveaux cas sont diagnostiqués chaque année.[2] Ces chiffres restent entourés d’incertitudes, faute de registre national exhaustif et en raison d’un sous-diagnostic persistant chez les personnes âgées ou présentant plusieurs pathologies.
La maladie associe plusieurs processus neuropathologiques. L’accumulation extracellulaire de peptides bêta-amyloïdes favorise la formation de plaques, tandis que les modifications de la protéine tau conduisent à des enchevêtrements neurofibrillaires à l’intérieur des neurones. Une perte synaptique, une neuro-inflammation et une dégénérescence progressive des réseaux cérébraux complètent ce tableau.
La présence de ces lésions ne se superpose pourtant pas toujours à l’intensité des symptômes. L’âge, la réserve cognitive, les atteintes vasculaires et la coexistence d’autres maladies neurodégénératives modulent l’expression clinique. Cette discordance explique pourquoi un résultat biologique ne peut être interprété sans tenir compte de l’histoire du patient.
La clinique reste la porte d’entrée du diagnostic
Le diagnostic de la maladie d’Alzheimer commence par l’entretien clinique. L’évolution des troubles, leur retentissement sur les activités quotidiennes, les observations des proches et la recherche de symptômes comportementaux constituent une trame qu’aucune prise de sang ne peut remplacer.
Les troubles de la mémoire épisodique sont fréquents, mais ils ne résument pas la maladie. Des difficultés langagières, visuospatiales ou exécutives peuvent dominer certains tableaux. Une désorientation, une perte d’autonomie financière, des erreurs médicamenteuses, une modification de la personnalité ou un retrait social doivent également attirer l’attention.
À l’inverse, une plainte mnésique isolée ne suffit pas à établir un diagnostic. Elle peut être liée à un trouble anxieux, à une dépression, à un trouble du sommeil, à un traitement médicamenteux ou à une autre pathologie neurologique.
L’évaluation cognitive et neuropsychologique objective les domaines atteints et précise leur sévérité. L’imagerie par résonance magnétique (IRM) recherche des lésions vasculaires, une tumeur, une hydrocéphalie, des séquelles traumatiques ou une atrophie compatible avec une maladie neurodégénérative.
Le bilan biologique conventionnel vise parallèlement à identifier des causes ou des facteurs aggravants accessibles à une prise en charge, tels qu’un trouble thyroïdien, une carence vitaminique, une anomalie métabolique ou un effet indésirable médicamenteux.[2]
Cette séquence clinique demeure déterminante. Un biomarqueur amyloïde positif atteste la présence d’une pathologie amyloïde, mais ne prouve pas que tous les symptômes observés lui soient imputables. Des dépôts peuvent être détectés chez une personne sans déficit cognitif manifeste, tandis que plusieurs lésions cérébrales coexistent fréquemment chez un même patient.
Une définition biologique qui transforme le diagnostic d’Alzheimer
Les critères publiés le 27 juin 2024 sous l’égide de l’Alzheimer’s Association proposent de définir la maladie d’Alzheimer à partir de sa biologie plutôt que du seul syndrome clinique.[3]
Selon ce cadre, l’anomalie d’un biomarqueur dit « Core 1 » suffisamment validé peut établir la présence d’une maladie biologique. Cette catégorie comprend notamment la tomographie par émission de positons (TEP) amyloïde, certains biomarqueurs du liquide cérébrospinal (LCS) et des biomarqueurs plasmatiques performants, parmi lesquels la protéine tau phosphorylée en position 217, ou p-tau217.
Cette évolution sépare deux dimensions autrefois étroitement mêlées. Le stade biologique décrit la présence et l’extension des processus pathologiques. Le stade clinique mesure, quant à lui, les symptômes et leur retentissement fonctionnel.
Une personne peut ainsi présenter une pathologie biologique sans trouble cognitif objectivable. À l’inverse, un syndrome démentiel peut être lié à une autre maladie ou à l’association de plusieurs mécanismes.
Ces critères ne constituent cependant pas un protocole clinique détaillé. Ils fournissent des principes destinés à rapprocher la recherche et les soins, mais ne définissent pas à eux seuls les indications d’un examen ou d’un traitement. Le groupe de travail déconseille d’ailleurs la recherche de biomarqueurs chez les personnes cognitivement indemnes en dehors des études observationnelles ou thérapeutiques.[3]
Cette réserve tient notamment à l’incertitude pronostique. La découverte d’un biomarqueur positif ne permet pas de prévoir avec précision si une personne asymptomatique développera des troubles, ni dans quel délai. Une telle information peut pourtant avoir des conséquences psychologiques, familiales, professionnelles ou assurantielles durables.
Les biomarqueurs sanguins rapprochent le diagnostic de la consultation
Jusqu’à récemment, la confirmation d’une pathologie Alzheimer reposait le plus souvent sur une ponction lombaire ou une TEP amyloïde. Ces examens restent des références, mais ils sont invasifs, coûteux ou inégalement disponibles. Les biomarqueurs sanguins pourraient alléger une partie du parcours diagnostique.
Les marqueurs plasmatiques les plus étudiés comprennent les formes phosphorylées de tau — p-tau217, p-tau181 et p-tau231 — ainsi que le rapport Aβ42/Aβ40. La p-tau217 apparaît particulièrement performante pour repérer une pathologie amyloïde chez des patients présentant un déficit cognitif.
Dès 2019, les recherches sur les marqueurs sanguins de la maladie d’Alzheimer annonçaient cette évolution, alors encore largement expérimentale.[12]
En octobre 2024, l’OMS a publié les caractéristiques souhaitables des futurs tests sanguins.[4] L’organisation insiste sur leur performance analytique, leur accessibilité et leur capacité à être utilisés dans des contextes sanitaires variés.
Elle souligne également que l’âge, les comorbidités, les traitements, les facteurs génétiques, la population étudiée et le stade de la maladie peuvent modifier l’interprétation d’un résultat.
Une recommandation clinique de l’Alzheimer’s Association, publiée le 29 juillet 2025, distingue deux usages chez les patients présentant un déficit cognitif objectivé et évalués dans une structure spécialisée.[5]
Un test atteignant plus de 90 % de sensibilité et plus de 75 % de spécificité peut servir au triage. Un résultat négatif rend alors la pathologie Alzheimer peu probable, tandis qu’un résultat positif doit être confirmé par une analyse du LCS ou une TEP amyloïde.
Lorsqu’un test dépasse 90 % de sensibilité et 90 % de spécificité, il peut, dans ce cadre précis, se substituer à un examen confirmatoire du LCS ou à une TEP amyloïde. Cette possibilité ne concerne ni tous les dosages disponibles, ni les personnes asymptomatiques.
La recommandation rappelle également qu’un biomarqueur sanguin ne doit pas être prescrit avant une évaluation clinique complète. Son résultat doit toujours être replacé dans la probabilité diagnostique préalable.
Deux techniques mesurant la p-tau217 ne sont pas automatiquement interchangeables. Leurs seuils, leurs performances réelles et leur sensibilité aux comorbidités peuvent différer. La valeur prédictive d’un test dépend aussi de la fréquence de la pathologie dans la population examinée.
Une étude publiée le 4 avril 2025 dans JAMA Neurology illustre cette difficulté.[6] Menée auprès de 969 participants issus des cohortes françaises MEMENTO et BALTAZAR, elle montre que les performances et les valeurs prédictives de la p-tau217 varient selon le profil cognitif.
La valeur prédictive positive atteignait 90 % chez les patients présentant un trouble cognitif léger au profil typique de maladie d’Alzheimer, contre 60 % chez les personnes consultant pour une plainte cognitive subjective. Les auteurs concluent qu’un phénotypage neuropsychologique détaillé doit accompagner l’interprétation du biomarqueur afin de limiter les faux positifs.
Les traitements anti-amyloïde rendent la confirmation biologique indispensable
L’essor des biomarqueurs est étroitement lié à l’arrivée de traitements ciblant les dépôts amyloïdes. Leur prescription suppose de démontrer que la cible biologique est effectivement présente. Un diagnostic seulement probable ne suffit plus.
Le lécanémab, commercialisé sous le nom Leqembi, dispose d’une autorisation valable dans l’Union européenne depuis le 15 avril 2025. Il est indiqué chez des adultes présentant un trouble cognitif léger ou une démence légère dus à la maladie d’Alzheimer, avec une pathologie amyloïde confirmée, et ne portant aucune copie ou une seule copie de l’allèle ε4 du gène de l’apolipoprotéine E (APOE ε4). Le traitement est administré par perfusion intraveineuse toutes les deux semaines.[8]
Dans la population correspondant à l’indication européenne, qui regroupait 1 521 participants, le score Clinical Dementia Rating–Sum of Boxes (CDR-SB) augmentait en moyenne de 1,217 point sous lécanémab contre 1,752 point sous placebo à 18 mois.
Une hausse plus faible traduit une progression moyenne moins prononcée des troubles cognitifs et fonctionnels, soit une différence de −0,535 point entre les groupes selon l’analyse de l’Agence européenne des médicaments.[8]
Le donanémab, commercialisé sous le nom Kisunla, a été autorisé dans l’Union européenne le 24 septembre 2025 pour une population comparable. Il est administré toutes les quatre semaines et la durée du traitement ne doit pas dépasser 18 mois.[9]
Dans la population correspondant à l’indication européenne, le score intégré Alzheimer’s Disease Rating Scale (iADRS) diminuait en moyenne de 10,82 points sous donanémab contre 13,47 points sous placebo après 76 semaines, soit une différence de 2,65 points en faveur du traitement.
L’iADRS s’étend de 0 à 144 points : plus le score est faible, plus la dégradation cognitive et fonctionnelle est marquée. Les deux groupes ont donc décliné au cours de l’essai, mais la baisse moyenne était moins prononcée sous donanémab.[9]
En mai 2023, les premiers résultats de phase 3 du donanémab avaient nourri l’espoir d’un changement thérapeutique.[13]
Les évaluations réglementaires ultérieures ont confirmé un ralentissement statistique du déclin chez certains patients. Elles ont aussi mis en évidence une quantité d’effet limitée, des contraintes organisationnelles lourdes et des risques neurologiques parfois graves.
Ces anticorps ne restaurent pas les capacités perdues et ne guérissent pas la maladie d’Alzheimer. Leur bénéfice moyen est mesuré sur des échelles cognitives et fonctionnelles dont la traduction dans la vie quotidienne demeure discutée.
La HAS maintient une ligne restrictive sur le remboursement
La position française s’est construite en deux étapes. Avant l’examen du remboursement de droit commun, la Haute Autorité de santé (HAS) a refusé l’accès précoce au lécanémab le 4 septembre 2025, après un avis de la Commission de la transparence daté du 27 août. Elle a ensuite refusé celui du donanémab le 12 mars 2026.[15][16]
L’accès précoce et le remboursement répondent à des procédures distinctes. Le premier vise à permettre l’utilisation anticipée d’un médicament dans une maladie grave ou invalidante lorsque les critères réglementaires sont remplis. Le second examine notamment le service médical rendu (SMR) et l’amélioration du service médical rendu (ASMR).
Pour le lécanémab, la Commission de la transparence a rendu, le 22 octobre 2025, un avis défavorable au remboursement, mis en ligne le 7 novembre. Elle a relevé une différence moyenne ajustée de −0,451 point sur l’échelle CDR-SB à 18 mois dans la population d’analyse principale.
« Quantité d’effet modeste considérée comme non cliniquement pertinente ».[10]
La HAS souligne également les limites méthodologiques de l’essai, le recul comparatif limité à 18 mois, l’absence de données robustes sur la qualité de vie et l’absence de démonstration d’une modification durable du cours de la maladie. Elle estime que le SMR est insuffisant pour justifier une prise en charge par la solidarité nationale.
Pour le donanémab, la Commission de la transparence a rendu un avis défavorable au remboursement le 6 mai 2026, mis en ligne le 26 mai.[11] La HAS reconnaît une supériorité statistique par rapport au placebo, mais estime que la quantité d’effet demeure modeste, que les données comparatives sont limitées à 76 semaines et que l’impact sur la qualité de vie ou l’évolution à long terme n’est pas suffisamment établi.
La différence minimale cliniquement pertinente, souvent désignée par l’acronyme anglais MCID, ne correspond pas à un seuil universel. Une revue systématique rapide publiée en 2024 montre que les estimations varient selon le stade de la maladie, l’échelle employée, la méthode statistique et la question posée.[17]
Le changement perceptible chez un patient ne se confond donc pas nécessairement avec la différence moyenne observée entre deux groupes dans un essai clinique.
La divergence entre l’Agence européenne des médicaments et la HAS ne constitue pas une contradiction réglementaire. L’agence européenne détermine si le rapport entre les bénéfices et les risques autorise la commercialisation dans une population définie. La HAS apprécie, de son côté, si le bénéfice clinique justifie une prise en charge financière en France.
Les ARIA constituent un risque fréquent et parfois grave
Les anomalies de l’imagerie liées à l’amyloïde, désignées par l’acronyme ARIA, ne sont pas exceptionnelles sous anticorps anti-amyloïde.
Elles comprennent les ARIA-E, principalement associées à un œdème ou à des épanchements, et les ARIA-H, qui recouvrent notamment les microhémorragies et la sidérose superficielle.
Dans la population éligible au lécanémab en Europe, une ARIA était observée chez 17 % des patients traités, contre 7 % sous placebo. Les ARIA-E concernaient 9 % des patients contre 1 % sous placebo, tandis que les ARIA-H touchaient 13 % des patients contre 7 %. Ces catégories peuvent se chevaucher.[8]
Des manifestations symptomatiques étaient rapportées chez 2 % des patients et des formes graves nécessitant une hospitalisation chez 0,4 %. Des hémorragies intracérébrales de plus d’un centimètre ont été observées chez 0,5 % des patients traités par lécanémab, contre 0,1 % sous placebo.
Sous donanémab, dans la population correspondant à l’indication européenne, environ 33 % des patients présentaient une ARIA, contre 13,5 % sous placebo. Les ARIA-E concernaient 20,6 % des patients traités et les ARIA-H 27,6 %.[9]
Des ARIA graves étaient rapportées chez 1,4 % des patients et trois décès liés à ces événements, soit environ 0,4 %, sont survenus dans l’étude pivot.
La plupart de ces anomalies demeurent asymptomatiques. Certaines provoquent néanmoins des céphalées, une confusion, des troubles visuels, des nausées, des difficultés à la marche, des crises convulsives ou des déficits neurologiques focaux.
Le risque augmente notamment avec le port de l’allèle APOE ε4, la présence initiale de microhémorragies, une sidérose superficielle ou des signes compatibles avec une angiopathie amyloïde cérébrale.
Pour le lécanémab, l’information produit européenne prévoit une IRM cérébrale réalisée dans les six mois précédant l’instauration, puis avant les 3e, 5e, 7e et 14e perfusions. Une imagerie supplémentaire est nécessaire en cas de symptômes évocateurs.[8]
Pour le donanémab, une IRM doit être réalisée dans les six mois précédant le traitement, puis avant les 2e, 3e, 4e et 7e doses. Un contrôle supplémentaire avant la 12e dose est prévu chez certains patients présentant des facteurs de risque.[9]
Le traitement doit donc être supervisé par une équipe formée à la détection et à la prise en charge des ARIA ainsi que des réactions liées aux perfusions.
L’anticoagulation réduit fortement le nombre de patients éligibles
L’anticoagulation constitue une limite pratique majeure. Le lécanémab comme le donanémab ne doivent pas être instaurés chez un patient recevant un traitement anticoagulant au long cours.
Si une anticoagulation devient nécessaire pendant le traitement, notamment en raison d’une embolie pulmonaire ou d’une thrombose artérielle, les perfusions doivent être interrompues. Elles ne peuvent être reprises que lorsque l’anticoagulant n’est plus médicalement indiqué.[8][9]
L’aspirine et les autres antiagrégants plaquettaires peuvent être maintenus selon les informations européennes, mais le risque hémorragique doit être évalué individuellement.
La thrombolyse appelle une vigilance renforcée, car une ARIA-E symptomatique peut provoquer un déficit neurologique focal et imiter un accident vasculaire cérébral ischémique. Avant toute thrombolyse chez un patient traité par un anticorps anti-amyloïde, l’équipe d’urgence doit donc rechercher ce traitement et envisager la possibilité d’une ARIA.
Le génotypage APOE intervient également dans la sélection des patients. Les personnes homozygotes pour l’allèle ε4 présentent un risque nettement accru d’ARIA et sont exclues des indications européennes.
Cette analyse révèle toutefois une information de susceptibilité génétique susceptible de concerner indirectement les apparentés. Elle suppose une information préalable, un consentement adapté et une interprétation prudente.
La HAS estime que ces exigences entraîneraient une « modification radicale du parcours de soins et de l’organisation des soins ».[10]
Le déploiement mobiliserait les consultations mémoire, les capacités d’IRM, les centres de perfusion, les laboratoires de génétique et les équipes capables d’assurer une réponse rapide en cas de complication.
L’analyse économique ne peut donc pas se limiter au prix du médicament. En l’absence de tarif français officiel publié pour ces traitements non remboursés, une estimation en euros serait prématurée.
Le coût réel du parcours comprendrait de toute façon la confirmation amyloïde, le génotypage, les perfusions répétées, les IRM de surveillance, le temps médical et la prise en charge éventuelle des effets indésirables.
Les associations de patients dénoncent une perte de chance
La lecture de la HAS n’épuise pas le débat. Le 28 mai 2026, France Alzheimer a qualifié le refus de remboursement du donanémab de décision « incompréhensible ».[18]
L’association considère que l’absence d’accès aux premiers traitements agissant sur un mécanisme biologique de la maladie crée une inégalité avec les pays qui ont choisi de les rendre disponibles.
Elle met également en avant la faiblesse des options thérapeutiques actuelles, la progression irréversible de la maladie et la fenêtre d’éligibilité limitée aux stades les plus précoces.
Dans cette perspective, quelques mois de déclin potentiellement retardé peuvent revêtir une valeur particulière pour les patients et leurs proches, même lorsque l’effet moyen est jugé modeste par une agence d’évaluation.
Cette position exprime les attentes des familles et une appréciation différente du bénéfice acceptable. Elle ne constitue pas une preuve supplémentaire d’efficacité.
Elle rappelle néanmoins que l’évaluation d’un traitement ne porte pas seulement sur une valeur statistique. Elle engage aussi la tolérance au risque, les préférences du patient, l’équité d’accès et la valeur accordée au maintien de l’autonomie.
La prévention reste le levier le plus accessible
La sophistication du diagnostic et l’arrivée des traitements anti-amyloïde ne doivent pas reléguer au second plan la prévention de la maladie d’Alzheimer et des autres démences.
La Commission permanente du Lancet estimait en 2024 que la fraction des démences attribuable à 14 facteurs potentiellement modifiables atteignait 45,3 %.[7]
Cette proportion ne signifie pas que 45,3 % des cas seraient évités avec certitude par des changements individuels. Elle correspond à une estimation théorique à l’échelle de la population, fondée sur des associations épidémiologiques et sur l’hypothèse d’une réduction des expositions recensées.
Les facteurs identifiés comprennent le faible niveau d’éducation, la perte auditive, l’hypertension, le tabagisme, l’obésité, la dépression, l’inactivité physique, le diabète, l’isolement social, la consommation excessive d’alcool, les traumatismes crâniens, la pollution atmosphérique, l’excès de cholestérol LDL et la perte visuelle non corrigée.
La prévention ne se limite donc pas à des conseils généraux sur le mode de vie. Elle passe par le dépistage et le traitement de l’hypertension et du diabète, la correction des troubles sensoriels, la réduction du tabagisme, la prévention des traumatismes, ainsi que le maintien d’activités physiques, cognitives et sociales.
Dès 2019, l’association entre plusieurs habitudes favorables et un risque moindre de démence faisait l’objet de présentations scientifiques.[14]
Les travaux plus récents renforcent cette approche multifactorielle, sans démontrer qu’une conduite isolée puisse empêcher la maladie.
Un diagnostic précoce doit déboucher sur une décision utile
L’élargissement des outils diagnostiques peut réduire l’errance de certains patients, faciliter l’accès aux essais cliniques et mieux caractériser l’origine des troubles.
Un résultat négatif peut également réorienter les investigations vers une autre maladie neurodégénérative, une pathologie vasculaire, un trouble psychiatrique ou une cause métabolique.
Diagnostiquer plus tôt ne constitue pourtant pas une fin en soi. L’examen doit répondre à une question clinique préalable et déboucher sur une décision : accompagnement du patient et de ses proches, traitement symptomatique lorsqu’il est indiqué, adaptation du domicile, évaluation de la conduite automobile, anticipation juridique et sociale ou discussion d’un traitement ciblé.
Les biomarqueurs sanguins pourraient éviter certaines ponctions lombaires ou TEP inutiles et raccourcir les délais. Ils pourraient aussi déplacer le goulot d’étranglement si chaque résultat positif ou intermédiaire impose une confirmation spécialisée.
Leur intérêt devra donc être apprécié à l’échelle du parcours diagnostique complet, et non à travers le seul coût du prélèvement.
La perspective thérapeutique renforce parallèlement les exigences du consentement. Le patient susceptible de recevoir un anticorps anti-amyloïde doit pouvoir mettre en balance un ralentissement moyen limité, des perfusions répétées, plusieurs IRM, le risque d’ARIA et la possibilité d’une interruption définitive du traitement.
Cette décision ne se réduit ni à un résultat biologique ni à un pourcentage d’efficacité.
Une médecine de précision encore inachevée
La maladie d’Alzheimer entre dans une phase où la biologie occupe une place sans précédent. Les biomarqueurs sanguins rapprochent la caractérisation de la pathologie de la consultation, tandis que le lécanémab et le donanémab rendent la confirmation biologique indispensable chez les patients susceptibles d’être traités.
Cette transition reste néanmoins inachevée. La valeur d’un biomarqueur dépend de la population testée, du contexte clinique, de la qualité analytique et de la décision susceptible d’en découler. La clinique conserve donc sa fonction de point de départ et de boussole.
Sans organisation adaptée, sans accompagnement après l’annonce et sans accès équitable aux examens confirmatoires, la précision biologique pourrait accroître les disparités territoriales.
Les patients éloignés des consultations mémoire, des plateaux de TEP, des laboratoires spécialisés ou des capacités d’IRM risquent d’être exclus d’un parcours déjà fortement sélectif.
Le changement ne réside donc pas seulement dans la possibilité de mesurer l’amyloïde ou la protéine tau dans le sang. Il tient à la capacité du système de santé à transformer cette information en une décision compréhensible, proportionnée et réellement utile au patient.
Références
-
Organisation mondiale de la santé, « Dementia », 3 juillet 2026.
-
INSERM, « Maladie d’Alzheimer », mise à jour du 13 octobre 2025.
-
Alzheimer’s & Dementia, « Revised criteria for diagnosis and staging of Alzheimer’s disease », 27 juin 2024.
-
Organisation mondiale de la santé, « WHO publishes preferred product characteristics for blood tests for Alzheimer disease », 23 octobre 2024.
-
Alzheimer’s & Dementia, « Alzheimer’s Association Clinical Practice Guideline on the use of blood-based biomarkers in the diagnostic workup of suspected Alzheimer’s disease within specialized care settings », 29 juillet 2025.
-
JAMA Neurology, « Cognitive Phenotyping and Interpretation of Alzheimer Blood Biomarkers », 4 avril 2025.
-
The Lancet, « Dementia prevention, intervention, and care: 2024 report of the Lancet standing Commission », 31 juillet 2024.
-
Agence européenne des médicaments, « Leqembi — information produit », autorisation européenne du 15 avril 2025, information produit actualisée en 2026.
-
Agence européenne des médicaments, « Kisunla — information produit », autorisation européenne du 24 septembre 2025, information produit actualisée en 2026.
-
Haute Autorité de santé, « LEQEMBI — lécanémab — Maladie d’Alzheimer », avis du 22 octobre 2025, mis en ligne le 7 novembre 2025.
-
Haute Autorité de santé, « KISUNLA — donanémab — Maladie d’Alzheimer », avis du 6 mai 2026, mis en ligne le 26 mai 2026.
-
Caducee.net, « Progrès dans la recherche mondiale de marqueurs sanguins de la maladie d’Alzheimer et d’autres formes de démence », 15 juillet 2019.
-
Caducee.net, « Alzheimer : le donanémab de Lilly réduit significativement le déclin cognitif et fonctionnel dans une étude de phase 3 », 4 mai 2023.
-
Caducee.net, « L’AAIC 2019 présente de nouvelles recherches sur le mode de vie et le risque de maladie d’Alzheimer », 18 juillet 2019.
-
Haute Autorité de santé, « LEQEMBI — lécanémab — refus d’accès précoce », 4 septembre 2025.
-
Haute Autorité de santé, « KISUNLA — donanémab — refus d’accès précoce », 12 mars 2026.
-
Alzheimer’s & Dementia, « Minimal clinically important difference in Alzheimer’s disease », 2024.
-
France Alzheimer, « Refus de remboursement du Kisunla : une décision incompréhensible ! », 28 mai 2026.
Descripteur MESH : Maladie , Maladie d'Alzheimer , Diagnostic